greene lab
virology and immunology
A Protective Host Response Gone Awry?
Home
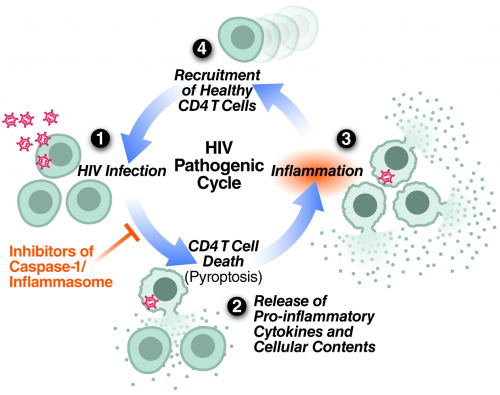
Repeating cycle of HIV abortive infection in resting CD4 T cells, pyroptotic cell death, inflammation and attraction of new cells to die proposed to centrally contribute to HIV pathogenesis and progression to AIDS.
Areas of Investigation
My laboratory seeks to better understand the interplay of human immunodeficiency virus (HIV) with its cellular host with the goal of providing new approaches for therapy and prevention. Currently we are exploring the molecular mechanisms by which HIV promotes CD4 T-cell death. The progressive loss of CD4 T cells in HIV-infected patients represents the fundamental problem that gives rise to acquired immunodeficiency syndrome (AIDS). Despite vigorous research over the last three decades, the precise mechanism underlying CD4 T-cell death during HIV infection has remained poorly understood. This topic has been highlighted as one of the five most important unanswered questions in HIV research.
Viral depletion of CD4 T cells has largely been studied in peripheral blood, the most accessible source of these cells. However, blood may not be the most relevant tissue for HIV research. Our comparative studies of lymphoid tissues and peripheral blood show that massive CD4 T-cell depletion by HIV preferentially occurs within lymphoid tissues, where more than 98% of CD4 T cells reside. We have specifically elected to explore the biological effects of HIV infection in an ex vivo human lymphoid aggregate culture (HLAC) system formed with fresh human tonsillar or splenic tissues. This system offers a unique experimental approach for modeling molecular and cellular events during HIV infection of human patients. HLACs can be infected with a small number of viral particles in the absence of added mitogens or cytokines, allowing analysis of HIV cytopathicity in a natural and preserved lymphoid microenvironment.
Infection of these cultures with HIV produces extensive loss of CD4 T cells, but >95% of the dying cells are not productively infected. Instead, we found that the dying cells are abortively infected with HIV. Due to viral nonpermissivity of these quiescent cells, the viral lifecycle attenuates during the chain elongation phase of reverse transcription, giving rise to incomplete cytosolic viral DNA transcripts. Importantly, these cells do not die because of a toxic action of these DNAs. Rather, death occurs as a consequence of a powerful, innate, defensive immune response launched by the host against these cytoplasmic viral DNA intermediates.
Recently we have shown that the mechanism of CD4 T-cell death is not “silent.” Instead, these abortively infected cells activate caspase-1 to produce a fiery form of programmed cell death termed pyroptosis. This death pathway causes significant inflammation, as the cells form pores in their plasma membrane through which their entire cytoplasmic contents are released. In bacterial infections, such release of inflammatory signals is thought to promote clearance by attracting more immune cells to help. However, in the case of HIV, a chronic viral infection, this strategy backfires. Instead of clearing the infection, proinflammatory signals released by pyroptosis attract more cells into the infected tissue to die and, in turn, produce chronic inflammation, creating a vicious pathogenic cycle that steadily depletes CD4 T cells.
Using unbiased proteomic and targeted biochemical approaches, as well as two independent methods of lentiviral short hairpin RNA-mediated gene knockdown in primary CD4 T cells, we have recently identified interferon-gamma–inducible protein 16 (IFI16) as a host DNA sensor required for CD4 T-cell death due to abortive HIV infection. Together, these findings provide insights into a key host pathway that plays a central role in CD4 T-cell depletion during disease progression to AIDS.
HIV latency represents a second active area of investigation in the laboratory. We are exploring the role of host factors and micro RNAs as regulators of latent HIV proviruses. Recently, we have identified mIR 155, a host factor that likely reinforces viral latency. We additionally identified TRIM32 as one of its key cellular targets and showed that TRIM32 activates NF-kB, an activator of latent HIV, via a novel mechanism involving direct ubiquitination of the IkBa, the inhibitor of NF-kB.
Significance
My laboratory seeks to understand the molecular basis for HIV pathogenesis. Through the insights gained from these studies, we hope to contribute to the development of new approaches to treat HIV-infected people and to prevent the spread of the virus. One new treatment strategy that we are currently pursuing is to restrain the hosts’ destructive response to HIV rather than the virus itself. Interestingly, an existing caspase-1 inhibitor—a drug already shown to be safe in humans—suppresses CD4 T-cell death and inflammation in HLACs. We are now planning a Phase II clinical trial to test its capacity to block pyroptosis in HIV-infected patients.
Approaches
Due to the broad scope of experimental questions, we employ a wide range of molecular, biochemical, cell biological and immunological techniques to study HIV pathogenesis. Individuals completing training in the laboratory routinely acquire expertise in all of these areas. Increasingly, we are utilizing an ex vivo human lymphoid aggregate culture (HLAC) system formed with fresh human tonsillar or splenic tissues. Our studies often take advantage of the outstanding core services offered at the Gladstone Institutes including: Mass Spectrometry, Immunology, Genomics, Flow Cytometry, Microscopy, and Histology.
Warner C. Greene, MD, PhD

Director and Senior Investigator, Michael Hulton Center for HIV Cure Research; Nick and Sue Hellmann Distinguished Professor; Professor of Medicine, Microbiology, and Immunology, University of California, San Francisco.
warner.greene@gladstone.ucsf.edu
415-734-4805