huang lab
neurological disease
Biological and Pathophysiological Functions of Apolipoprotein E
Home
Areas of Investigation
Research in our laboratory focuses on the biological and pathophysiological functions of apolipoprotein (apo) E. Long-term goals of our research are to understand the molecular and cellular mechanisms by which apoE4 increases the risk of Alzheimer’s disease (AD) and related neurodegenerative disorders and to develop therapeutic strategies.
Significance
Human apoE has three common isoforms (apoE2, apoE3 and apoE4) with different effects on lipid and neuronal homeostasis. ApoE4, the major known genetic risk factor for AD, increases the occurrence and lowers the age of onset of the sporadic and some familial forms of AD.
In most clinical studies, apoE4 carriers account for 65–80% of all AD cases, highlighting the importance of apoE4 in AD pathogenesis. Emerging data suggest that apoE4, with its multiple cellular origins and multiple structural and biophysical properties, contributes to AD by interacting with different factors through various pathways. However, the exact mechanisms underlying apoE4’s contribution to AD are still unclear.
Approaches
We use neuronal cultures, iPS cell technology, and transgenic and gene-targeted mouse models to study the different effects of apoE3 and apoE4 on cell signaling pathways and cytoskeletal structure and function at molecular, cellular and behavioral levels. We also use these approaches to develop and evaluate novel treatment strategies for apoE4-related neurodegenerative disorders.
Contributions
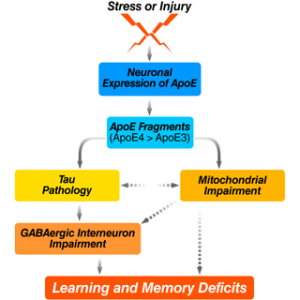
Studies from our laboratory have demonstrated a biological event that could play a major role in apo-related neuropathology. Specifically, apoE is subject to cleavage by a neuro-specific chymotrypsin-like serine protease that generates bioactive carboxyl-terminal-truncated forms of apoE (Brecht et al., J. Neurosci. 2004, 24:2527–2534; Harris et al., Proc. Natl. Acad. Sci. USA. 2003., 100:10966–10971). ApoE4 is more susceptible to cleavage than apoE3. The apoE fragments are found at higher levels in the brains of AD patients than in age- and sex-matched controls. When expressed in cultured neuronal cells or added exogenously to the cultures, the truncated apoE4 is neurotoxic leading to cytoskeletal disruption and mitochondrial dysfunction and finally cell death (Huang et al, Proc. Natl. Acad. Sci. USA. 2001, 98:8838–8843; Chang et al, Proc. Natl Acad. Sci. USA. 2001, 102:18694–18699). Furthermore, expression of the carboxyl-terminal-truncated apoE4 causes AD-like neurodegeneration and behavioral deficits in transgenic mice (Harris et al, Proc. Natl. Acad. Sci. USA. 2003, 100:10966–10971). Since we have demonstrated that apoE is synthesized by neurons under diverse physiological or pathological conditions (Harris et al, J. Bio. Chem. 2004, 279:3862–3868; Xu et al, J. Neurosci. 2006, 26:4985–4994; Xu et al, J. Neurosci. 2008, 28:1452–1459), this cleavage could represent an early event in apoE4-related neuropathology (Harris et al, J. Biol. Chem. 2004, 279:44795–44801).
Based on these in vitro and in vivo observations, we hypothesize that apoE4 produced in neurons in response to stress or injury (e.g. aging, Aß toxicity, brain trauma, or oxidative stress) is uniquely susceptible to proteolytic cleavage by an apoE cleaving enzyme (AECE) and that the resulting bioactive carboxyl-terminal-truncated fragments, probably together with other AD related factors (e.g., Aß) induce neuropathology and associated behavioral deficits. Thus, AECE represents a new therapeutic target for AD drug development. In addition, protection of the apoE4 fragment interaction with cytoskeletal components of mitochondria might also be beneficial for treatment or prevention from AD.
Recently, we also demonstrated that apoE4 knock-in (ApoE4KI) mice have a significant age-dependent decrease in hilar GABAergic interneurons that correlates with the extent of apoE4-induced impairment of adult hippocampal neurogenesis and learning and memory deficits (Li et al, Cell Stem Cell. 2009, 5:634–645; Andrews-Zwilling et al, J. Neurosci. 2010, in press). In neurotoxic apoE4 fragment transgenic mice, the interneuron loss was even more pronounced and correlated with the extent of learning and memory deficits. The interneuron loss and learning and memory deficits in these mice were prevented by tau removal, and the prevention was abolished by blocking GABA signaling with picrotoxin. In both groups of mice, the GABA receptor potentiator pentobarbital rescued the hippocampal neurogenesis impairment and learning and memory deficits. These findings strongly suggest that apoE4 causes age- and tau-dependent hilar GABAergic interneuron impairment, leading to adult hippocampal neurogenesis impairment and learning and memory deficits in mice.
Some Questions Addressed in Ongoing Studies:
- Why is apoE4 more susceptible than apoE3 to proteolysis?
- Which enzyme or enzymes are responsible for apoE cleavage?
- How do apoE4 fragments cause cytoskeletal disruption and mitochondrial dysfunction?
- Can we protect mice or humans from apoE4-related neurodegeneration by blocking its proteolysis?
- How is apoE expression regulated in neurons?
Yadong Huang, MD, PhD

Senior Investigator
Gladstone Institute of Neurological Disease
Professor of Neurology and Pathology
University of California, San Francisco
yhuang@gladstone.ucsf.edu
415-734-2000
415-355-0824 (F)