nakamura lab
neurological disease
Understanding and Treating Mitochondrial and Metabolic Dysfunction in Neurodegenerative Diseases


Home
Areas of Investigation
In our laboratory, we have two broad, intertwined objectives. The first is to gain insight into the normal physiology of mitochondria and glucose metabolism in the brain, with a particular focus on bioenergetics, mitochondrial dynamics, and quality control in neurons. The second is to understand how disrupting mitochondrial function and metabolism contributes to neurodegenerative diseases, especially Parkinson’s disease (PD), Alzheimer’s disease (AD), and mitochondrial disorders, and to use these insights to develop new approaches to target energy metabolism therapeutically.
Significance
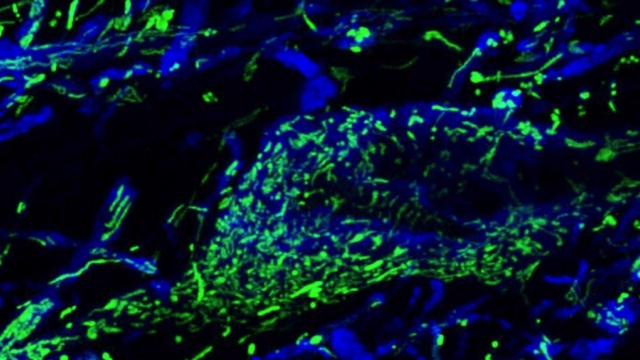
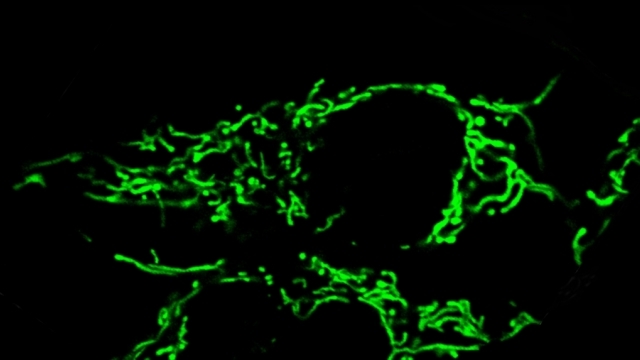
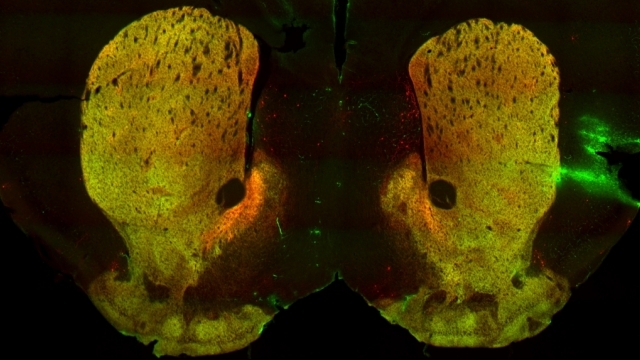
Mitochondria are dynamic organelles that frequently undergo fusion and fission. They are important in multiple cellular functions—including energy production—and are ultimately degraded. However, many aspects of mitochondrial behavior and function are not understood, especially in the brain and at nerve terminals. In addition, changes in mitochondria and metabolism contribute to, and sometimes even initiate, neurodegeneration, but the underlying mechanisms—and the mitochondrial changes themselves—are poorly characterized. What is clear is that different types of neurons require distinct mitochondrial functions and proteins, and that loss of these functions causes selective neuronal loss in a range of neurological disorders, including Charcot-Marie-Tooth disease, optic atrophy, mitochondrial diseases, and PD. Therefore, to understand how mitochondria contribute to neurodegeneration, they must be studied in susceptible neurons. Moreover, it is critical to understand mitochondrial biology in axons. Most mitochondria in neurons are believed to be in axons, and these mitochondria have different intrinsic properties and bioenergetic environments than those in other subcellular compartments. Axons are also lost early in many diseases that involve energy failure, and key pathogenic proteins such as α-synuclein and tau concentrate in axons. Thus, by understanding the normal behaviors and functions of axonal mitochondria in susceptible neuron types, we can begin to unravel how mitochondrial biology is disrupted in disease and, ultimately, design new mitochondria-based therapies.
Approaches
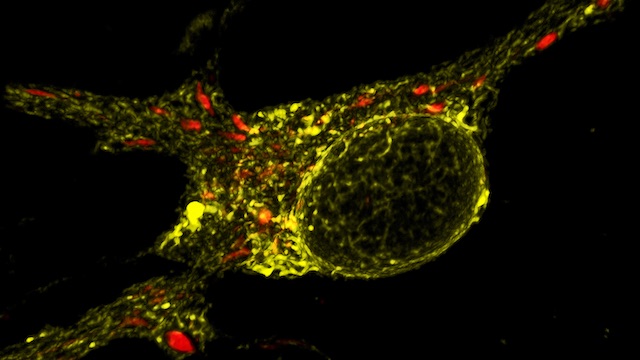
To study mitochondrial biology and metabolism in the brain, we use an array of imaging, cell engineering, metabolomic and biochemical approaches, in conjunction with model organisms. We visualize mitochondria in real time with targeted fluorescent probes, and we image mitochondrial bioenergetics, movement, and turnover in mammalian cells, including primary rodent and human neurons and their synapses. Our in vivo investigations utilize transgenic mouse models and genetically modified viral vectors. With this platform, we can investigate how mutations in patients with PD or AD disrupt metabolism and produce neurodegeneration. To establish mechanisms, we also use in vitro model systems with recombinant proteins and purified mitochondria or artificial membranes.
Contributions
Understanding and targeting energy failure in neurodegeneration. We developed new live-imaging assays to measure mitochondria-derived ATP in individual synaptic boutons, and used these assays to begin to define how much ATP is needed in different phases of the synaptic vesicle cycle. We found that endocytosis has particularly high ATP requirements (Pathak et al., JBC, 2015, selected by JBC editors as the top paper from bioenergetics in 2015). We also found that ATP is rapidly dispersed in axons, thereby maintaining nearly normal levels of ATP even in boutons lacking mitochondria. As a result, boutons without mitochondria have the same capacity for synaptic vesicle cycling as those with mitochondria. We also found that loss of a key respiratory subunit implicated in Leigh’s disease markedly decreases mitochondria-derived ATP in axons, resulting in a failure of synaptic vesicle cycling. Thus, mitochondria-based energy failure can occur and be detected in individual neurons in a model of neurodegeneration. However, we also found that ATP depletion does not occur under the high-glucose conditions typically used to study neurodegeneration, because neurons compensate by producing energy through glycolysis. Our findings thus highlight a key flaw in the approach currently used to study mitochondrial defects in culture; we instead offer a new and more sensitive approach to study mitochondria-based energy failure in neurodegeneration.
To identify genes that regulate ATP, we developed a novel high throughput genetic screen combining a FRET-based ATP biosensor, CRISPR technology and flow cytometry, enabling the fractionation of individual cells on the basis of real-time intracellular ATP levels. We first focused on a subset of mitochondrial genes known to impact ATP; we showed that mitochondrial ribosomal proteins are by far the most critical genes for maintaining ATP levels when mitochondria produce ATP, and identified genetic defects responsive to energy-based therapies (Mendelsohn et al., PLoS Biol, 2018). Remarkably, loss of these mitochondrial ribosomal proteins did not decrease ATP under glycolytic conditions. Instead, in many cases, it actually protected ATP levels, reflecting how cells forced to rely on glycolysis can adapt to have a more robust glycolytic capacity than controls.
These studies led to a major effort to identify new genetic pathways that regulate energy levels, forming the basis for the first comprehensive meta¬bolic map of mitochondrial genes and pathways that regulate cellular energy levels (Bennet et al., Nat. Commun., 2020). In this work, we greatly expanded the scope of our initial analysis, screening the entire genome with both CRISPRi and CRISPRa in three distinct metabolic conditions to interrogate substrate specificity of gene effects. Our study thus defined the “ATPome,” a comprehensive database of the effects of increasing or decreasing the expression of every gene on cellular ATP levels. On a systems level, this work defined broad mechanisms by which genes regulate ATP levels, including identifying hexokinase-2 activity as one of the greatest energy consumers in the genome. This work also identified central and opposing roles for the HIF1 pathway and mitochondrial translation and respiration in driving metabolism: suppression of each of these pathways effectively stimulates energy production by the other. Remarkably, we found that the HIF1 pathway strongly controls energy levels under non-hypoxic conditions, where it has been widely believed to be inactive.
Prior to this work, there were only anecdotal examples of how to increase ATP, and even then, the impact on ATP was usually limited to only a single substrate condition. By defining the ATPome, we believe our work opens new avenues to understand the molecular mechanisms of energy control, and reveals novel therapeutic strategies and targets for diseases of energy dysregulation, including neurodegeneration and cancer.
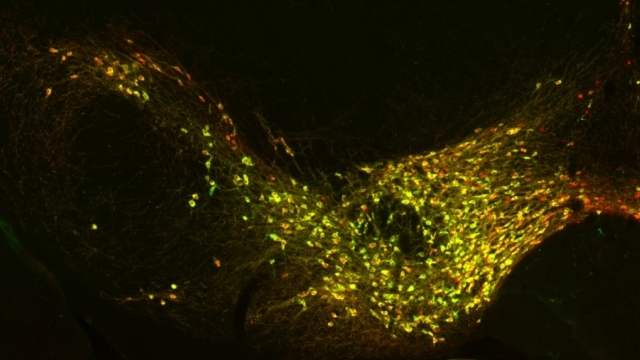
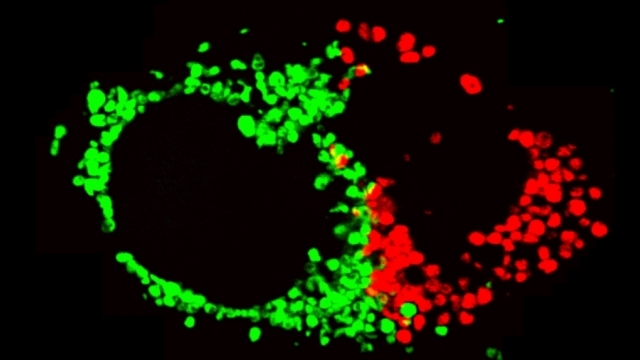
Mitochondrial fission at the synapse in susceptible neuronal populations. In separate work, we studied the normal functions of mitochondrial fission, a process whose disruption may contribute to both PD and AD. We established new animal models in which the central mitochondrial fission protein—dynamin-related protein 1 (Drp1)—was deleted in dopamine (DA) neurons, which are susceptible in PD (Berthet et al., J Neurosci, 2014) and in CA1 and other hippocampal neurons, which are susceptible in AD (Shields et al., Cell Death Dis, 2015). We found that Drp1 is critical for targeting mitochondria to the nerve terminal in DA neurons. We also discovered that disrupting mitochondrial fission causes the preferential death of nigrostriatal DA neurons but does not affect a subset of DA neurons with characteristic electrophysiologic properties in the adjacent ventral tegmental area. Interestingly, in contrast to nigrostriatal DA neurons, most CA1 neurons can survive without Drp1. Nonetheless, CA1 hippocampal neurons require mitochondrial fission for normal function, since loss of Drp1 compromises both pre- and postsynaptic functions, at least in part because the intrinsic respiratory function of axonal mitochondria lacking Drp1 is compromised. Mitochondrial fission is thus required to the maintain energy levels needed to support synaptic transmission, and disrupting mitochondrial fission in CA1 hippocampal neurons compromises synaptic function and may predispose to neurodegeneration.
α-Synuclein and mitochondria. Using optical FRET reporters for synuclein conformation, we found that the central PD protein α-synuclein preferentially binds to mitochondria versus other organelles due its high affinity for the acidic phospholipid cardiolipin, which is enriched in mitochondria (Nakamura et al., J Neurosci, 2008). We also discovered that increasing synuclein produces a dramatic and specific increase in mitochondrial fragmentation in a range of cell types, including DA neurons in transgenic models of PD. The mechanism involves a direct interaction of synuclein with mitochondrial membranes, and is mediated by a small oligomeric form of synuclein (Nakamura et al., JBC, 2011, selected by JBC editors as the top paper in neurobiology in 2011). These findings reveal a new function for synuclein in regulating mitochondrial morphology and establish a potential mechanism by which synuclein may produce degeneration in PD.
Questions Addressed in Ongoing Studies
• Why are substantia nigra DA neurons intrinsically vulnerable to mitochondrial stressors?
• How do PD proteins disrupt mitochondrial function and produce neurodegeneration?
• How do changes in glucose and energy metabolism contribute to the pathogenesis of AD?
• How do chronic changes in neural activity influence neurodegeneration?
• How and why are mitochondria turned over?
• How can we restore or even boost energy levels in cells, and will this protect against neurodegeneration?
Ken Nakamura, MD PhD

Senior Investigator
Gladstone Institute of
Gladstone Institute of
Neurological Disease
Professor
Professor
of Neurology
University of California,
San Francisco
University of California,
San Francisco
415-734-2000