palop lab
neurological disease
Mechanisms of Network and Interneuron Dysfunction in Alzheimer's Disease
Home
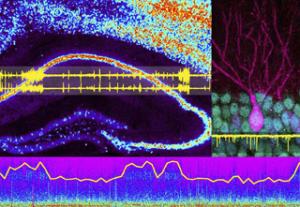
Research Interest
The Palop Laboratory seeks to understand the neuronal processes underlying cognitive impairments in neurodegenerative disorders, such as Alzheimer’s disease (AD), and in neuropsychiatric conditions associated with abnormal synchronization of neuronal networks, such as schizophrenia, autism, and epilepsy. We aim to identify molecular, circuit, and network mechanisms of cognitive dysfunction and to develop novel therapeutic approaches to restore brain functions in AD and related disorders. We are particularly focused on understanding the role of impaired inhibitory interneurons in network hypersynchrony, altered oscillatory brain rhythms, and cognitive dysfunction in AD.
To study these complex diseases, the Palop Lab primarily uses mouse models that recapitulate key aspects of the cognitive dysfunction and pathology of these conditions to dissect network and circuits mechanisms of brain dysfunction in mouse models of AD. We use electroencephalography (EEG), local field potentials (LFP), and single-unit recordings to assess neuron activity in vivo, optogenetic approaches to modulate interneuron function in vivo, genetic and pharmacological manipulations to manipulate specific pathways in vivo, and behavioral assessment to determine the cognitive consequences of our mechanistic interventions
Jorge J. Palop, PhD

Assistant Investigator
Gladstone Institute of Neurological Disease
Assistant Professor
Department of Neurology
University of California, San Francisco
jpalop@gladstone.ucsf.edu
415-734-2661
415-355-0824 (F)