mucke lab
neurological disease
Memory Loss and Major Neurological Deficits In Alzheimer's Disease and Related Neurodegenerative Disorders
Home
Areas of investigation
We study processes that result in memory loss and other major neurological deficits, with an emphasis on Alzheimer's disease (AD) and related neurodegenerative disorders. Our long-term goal is to advance the understanding of the healthy and the diseased central nervous system to a point where rational strategies can be developed for the prevention and cure of these conditions.
Significance
Molecules similar to those involved in neurodegenerative diseases are highly expressed in the nervous system of diverse species and appear to function in learning, synaptic plasticity, and regeneration.
We are particularly curious about the roles of amyloid proteins, tau and apolipoprotein E in AD, and about α-synuclein in AD and Parkinson's disease (PD). AD and PD are the most frequent neurodegenerative disorders. They erode people's ability to think and control their movements, two of the most critical and intriguing functions of the central nervous system. Both conditions are on the rise and neither can be prevented or cured. These facts underline the significance and urgency of our research efforts.
Approaches
We use transgenic mouse models and neural cultures to study potential pathogenic factors and pathways at the molecular, cellular, network, and behavioral level. Mouse models are also used to develop and evaluate novel treatment strategies. Their relevance is assessed through comparative studies of humans and postmortem tissues in collaboration with clinical programs.
Contributions
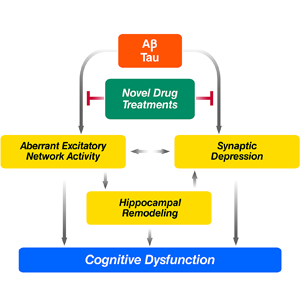
In AD-related transgenic models, we discovered that amyloid-β peptides (Aβ) can damage synapses and disrupt neural memory circuits independent of their deposition into the visible amyloid plaques that form in AD brains. The plaque-independent toxicity of Aβ was inhibited by apolipoprotein E3, but not E4, which may relate to the differential effects of these molecules on AD risk and age of onset. Pathogenic interactions between Aβ and α-synuclein worsened cognitive and motor deficits in doubly transgenic mice, a finding of potential relevance to the frequent overlap between AD and PD. Most recently, we discovered that neural network activity in AD-related mouse models fluctuates between abnormal excitation (epilepsy-like) and abnormal inhibition. Remarkably, reducing the protein tau effectively prevented these alterations as well as Aβ-induced cognitive deficits. Ongoing studies aim to determine whether such network dysfunction also contributes to cognitive deficits in AD.
Some questions addressed in ongoing studies
- How does Aβ affect synaptic function and neuronal survival?
- How does tau reduction make the brain resistant against Aβ-induced deficits?
- Can the beneficial effect of tau reduction be exploited therapeutically?
- Which drugs can block the aberrant network activity that Aβ triggers?
- Will these drugs also normalize cognitive functions and prevent neurological decline in AD?
- What can the selective vulnerability of specific neuronal populations to different neurodegenerative disorders teach us about the uniqueness of the affected cells and the pathogenic cascades involved?
Lennart Mucke, MD

Director and Senior Investigator
Gladstone Institute of
Neurological Disease
Professor of
Neurology and Neuroscience
University of California,
San Francisco
Email
415-734-2504 (T)
415-355-0131 (F)