huang lab
neurological disease
ApoE Research
Aβ-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer's disease
Overview
Human apolipoprotein (apo) E has three common isoforms (apoE2, apoE3, and apoE4) with different effects on lipid and neuronal homeostasis. ApoE4, the major known genetic risk factor for Alzheimer's disease (AD), increases the occurrence and lowers the age of onset of the sporadic and some familial forms of AD. In most clinical studies, apoE4 carriers account for 65–80% of all AD cases, highlighting the importance of apoE4 in AD pathogenesis. Emerging data suggest that apoE4, with its multiple cellular origins and multiple structural and biophysical properties, contributes to AD by interacting with different factors through various pathways, some of which are dependent on amyloid-β (Aβ). Although the Aβ-dependent roles of apoE4 in AD pathogenesis have been widely studied, the Aβ-independent roles of apoE4 have drawn less attention and have been understudied. Independently of Aβ, apoE4 has detrimental effects on neuronal plasticity, undergoes aberrant proteolysis that generates neurotoxic fragments, stimulates tau phosphorylation and disrupts the cytoskeleton, and impairs mitochondrial function.
| ApoE polymorphism and Alzheimer's disease ApoE, a 299–amino acid glycoprotein with a molecular weight of 34,200, is a polymorphic protein (Box 1) [1]. Its three common isoforms, apoE2, apoE3, and apoE4 [1-4], are all products of the same gene, which exists as three alleles (ε2, ε3, and ε4) at a single gene locus [5]. |
|
| Therefore, three homozygous (apoE2/2, E3/3, and E4/4) and three heterozygous (apoE3/2, E4/3, and E4/2) phenotypes occur in humans. In almost all populations, the apoE3/3 phenotype is the most common (typically ~50–70% of the population), and the ε3 allele accounts for the vast majority of the apoE gene pool (typically ~70–80%). The ε4 allele accounts for 10–15%, and the ε2 allele for 5–10%. The molecular basis for apoE polymorphism has been elucidated by amino acid sequence analysis [6-8]. The three isoforms differ from one another at one or two amino acid residues. ApoE3 has Cys-112 and Arg-158, whereas apoE4 has arginines at both positions, and apoE2 has cysteines (Box 1). | |
|
Box 1
ApoE: polymorphism, structural domains, and biophysical properties
|
|
| The sequence and structural differences in apoE isoforms have significant functional consequences [7,9]. ApoE4 is the major known genetic risk factor for AD (Boxes 1 and 2) [10,11]. In most clinical studies, apoE4 carriers account for 65–80% of all AD cases, highlighting the importance of apoE4 in AD pathogenesis [12]. | |||
|
ApoE4 is associated with increased formation of amyloid plaques and neurofibrillary tangles (NFTs) [13-16] that are characteristic of AD. The apoE4 allele is overrepresented in late-onset familial AD patients (0.50) compared with age- and sex-matched controls (0.16) [15]. The association of the apoE4 allele with late-onset familial AD has been confirmed in several populations and has been extended to late-onset sporadic AD [11]. Furthermore, apoE4 has a gene dose effect on the risk and age of onset of AD [10].
|
|
||
| As the number of apoE4 alleles increases from 0 to 1 to 2, the risk of developing late-onset familial AD increases from 20% to 90%, and the mean age of onset decreases from 84 to 68 years [10]. On the other hand, apoE2 may protect against AD development in some populations [17,18]. In addition, the apoE4 allele is also associated with poor clinical outcome in patients with acute head trauma [19-25]. Recently, apoE4 has also been suggested to be associated with other types of neurodegenerative disorders. ApoE4 may affect the age of onset, progression, or severity of stroke [26,27], Parkinson's disease [28-31], multiple sclerosis [32,33], and amyotrophic lateral sclerosis [34-38]. | |||
Structure, biophysical properties, and functions of apoE in neurobiology
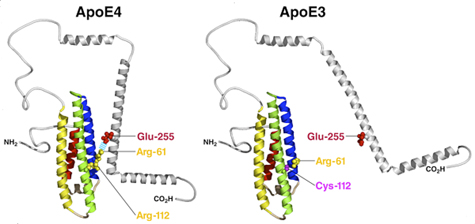
ApoE has two structural domains: the amino-terminal domain (amino acids 1–191), which contains the receptor binding region (amino acids 135–150), and the carboxyl-terminal domain (amino acids 223–299), which contains the lipid binding region (amino acids 241–272) [9]. The two domains are linked by a structurally flexible hinge region (amino acids 192–222) [9]. Interaction between the carboxyl- and amino-terminal domains, called domain interaction, is a unique biophysical property of apoE4 (Figure 1) [39,40]. In apoE4, Arg-112 reorients the side chain of Arg-61 in the amino-terminal domain from the position it occupies in apoE2 and apoE3, allowing it to form a salt bridge with Glu-255 in the carboxyl-terminal domain (Figure 1) [39,40]. In apoE2 and apoE3, Arg-61 has a different conformation, and domain interaction does not occur (Figure 1) [39,40]. Only human apoE has Arg-61; the 17 other species in which the apoE gene has been sequenced all have Thr-61 [9]. Mutation of Arg-61 to threonine or Glu-255 to alanine in apoE4 prevents domain interaction and converts apoE4 to an apoE3-like molecule [39,40]. ApoE4 domain interaction also occurs in living neuronal cells in culture [41]. Intramolecular domain interaction is responsible for apoE4's susceptibility to proteolysis (see below) [42,43] and for apoE4-caused astrocytic dysfunction [44,45].
Interestingly, mouse apoE contains arginine at a position equivalent to 112 in human apoE, but it lacks the critical Arg-61 that mediates domain interaction. Instead, mouse apoE contains Thr-61 [9]. Thus, mouse apoE lacks domain interaction and is functionally equivalent to human apoE3. Since mouse apoE contains the equivalent of Glu-255, replacing Thr-61 with arginine introduces domain interaction in gene-targeted mice [46].
|
ApoE has important and diverse roles in neurobiology [43,47-50]. The brain is second only to the liver in the abundance of apoE mRNA [51]. Brain apoE is synthesized and secreted primarily by astrocytes [52,53], but some neurons do so as well [54-63]. In some neurons, brain injury induces apoE expression [64]. ApoE-containing high density lipoproteins are found in cerebrospinal fluid, where they account for the majority of lipoproteins [65]. Moreover, apoE levels increase 250–350-fold in response to peripheral nerve injury in a rat model [66-69]. Injury-induced accumulation of apoE also occurs in the central nervous system (CNS), although to a lesser extent [70].
|
|
ApoE appears to take up lipids generated after neuronal degeneration and redistributes them to cells requiring lipids for proliferation, membrane repair, or remyelination of new axons [70,71]. ApoE may protect against motor and cognitive defects due to acute head injury [72] or stroke [27]. Increased production of apoE3, but not apoE4, in the hippocampus stimulates repair of local lesion-induced damage [73]. Moreover, synaptic and dendritic alterations and significant learning deficits, all of which have been associated with local neurite remodeling, are observed in apoE-deficient mice [74] and apoE4 transgenic mice [75-80]. Notably, apoE3, but not apoE4, protects against excitotoxin-induced neuronal damage in mice [75].
ApoE4 causes neuronal and behavioral deficits in the absence of Aβ accumulation in transgenic mice
Several transgenic mouse lines expressing apoE3 or apoE4 have been established. The neuron-specific enolase (NSE) promoter has been used to express human apoE3 or apoE4 at similar levels in neurons of transgenic mice lacking endogenous mouse apoE [75,78]. NSE-apoE4 mice show impairments in a water maze test and in vertical exploratory behavior not observed in NSE-apoE3 mice or wildtype controls. These impairments increase with age and, like in humans, are observed primarily in female apoE4 transgenic mice, suggesting that human apoE isoforms differ in their effects on brain functionin vivo and that the susceptibility to apoE4-induced deficits is critically influenced by age and gender [75,78]. Morphological studies of these transgenic mouse lines demonstrate that human apoE3 prevents the age-dependent neurodegeneration seen in apoE-null mice and prevents kainic acid–induced neurodegeneration [75]. Human apoE4 is not protective [75]. Transgenic mice expressing apoE4 in astrocytes have impairment of working memory, despite the absence of significant neuropathological changes in the brain [81]. Furthermore, human apoE4 knock-in mice, in which the mouse apoE gene is replaced with the human apoE gene, develop spatial learning and memory deficits not seen in human apoE3 knock-in mice [78,79,81,82]. Since Aβ does not accumulate in any of these apoE isoform transgenic mouse models, these data strongly suggest Aβ-independent roles of apoE4 in causing neuronal and behavioral deficitsin vivo.
ApoE4 impairs neuronal plasticity
In the presence of a source of lipids, apoE3 and apoE4 have markedly different effects on neurite extension [83-90]. In both dorsal root ganglion neurons and Neuro-2a cells in culture, apoE3 plus β-very low density lipoprotein (VLDL) significantly stimulates neurite extension, whereas apoE4 plus β-VLDL markedly inhibits neurite branching and extension [83,84,88]. In addition, astrocyte-derived apoE3, but not apoE4, stimulates neurite outgrowth of rat hippocampal neurons [90]. Furthermore, apoE3-transfected Neuro-2a cells grown in medium containing β-VLDL or high density lipoprotein from cerebral spinal fluid show greater neurite extension than apoE4-transfected Neuro-2a cells [91]. Finally, apoE3 stimulates, but apoE4 inhibits, neuronal sprouting in anin vitro mouse organotypic hippocampal slice culture system derived from transgenic mice expressing apoE3 or apoE4 [92]. ApoE4-associated inhibition of neurite extension is probably due to its effect on microtubule stability [84] and is mediated by cell-surface lipoprotein receptors, specifically the heparan sulfate proteoglycans/low density lipoprotein receptor pathway [83,88, 91]. Notably, apoE receptors mediate neurite outgrowth by activating the Erk pathway in primary neuronal cultures [93].
ApoE4 also impairs synaptogenesisin vivoin apoE transgenic and gene-targeted mice andin vitroin primary neuronal cultures. As compared to apoE3, apoE4 decreases dendritic spine density in transgenic and gene-targeted mice [94,95]. In rat primary cortical neuronal cultures, apoE4 and its fragment decrease the density of dendritic spines [96]. Interestingly, rosiglitazone, an insulin sensitizer and mitochondrial activator, rescues this loss of dendritic spines [96], suggesting the involvement of apoE4-caused mitochondrial impairment in the detrimental effect of apoE4 and its fragment on synaptogenesis.
ApoE4 also impairs adult hippocampal neurogenesis [97,98]. Adult mouse neural stem cells express apoE [98]. ApoE knockout mice have significantly less hippocampal neurogenesis, but significantly more astrogenesis, than wildtype mice due to decreased Noggin expression in neural stem cells [98]. In contrast, neuronal maturation in apoE4 knock-in mice is impaired due to reduced survival and function of GABAergic interneurons in the hilus of the hippocampus, and a GABAA receptor potentiator rescues the apoE4-associated decrease in hippocampal neurogenesis [98]. Thus, apoE contributes to adult hippocampal neurogenesis, and apoE4 impairs GABAergic input to newborn neurons, leading to decreased neurogenesis [98]. Interestingly, exercise, which stimulates hippocampal neurogenesis, improves cognition and hippocampal plasticity in apoE4 transgenic mice [99].
ApoE4 proteolysis in neurons and neurotoxicity
Initially, apoE was thought to be synthesized by astrocytes, oligodendrocytes, activated microglia, and ependymal layer cells in the brain [52,100]. However, CNS neurons also express apoE, albeit at lower levels than astrocytes [54-59,61,101,102]. Both apoE protein and mRNA are found in cortical and hippocampal neurons in humans [61] and in transgenic mice expressing human apoE under the control of the human apoE promoter [103]. In rats treated with kainic acid, apoE expression is induced in hippocampal neurons that survive excitotoxic stress, as determined by both in situ hybridization and anti-apoE immunohistochemistry [64]. Neuronal expression of apoE can be induced in human brains after cerebral infarction [104]. In EGFPapoE reporter mice, CNS neurons express apoE in response to excitotoxic injury [63]. ApoE is also expressed in primary cultured human CNS neurons [60] and in many human neuronal cell lines, including SY-5Y, Kelly, and NT2 cells [105,106].
|
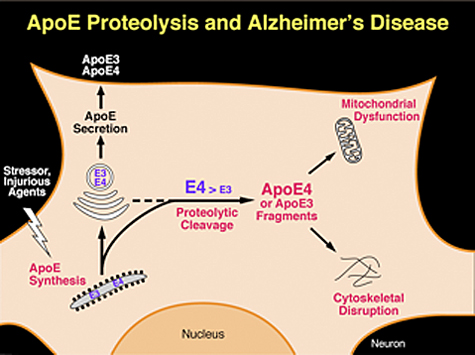
Neuronal apoE4 is more susceptible to proteolytic cleavage than neuronal apoE3, as determinedin vitro in transfected neuronal cells andin vivoin transgenic mice expressing apoE3 or apoE4 and by incubating recombinant apoE3 or apoE4 with partially purified apoE cleaving enzyme, a chymotrypsin-like serine protease, from neurons (Figure 2) [107-109]. ApoE fragments are present at much higher levels in the brains of AD patients than in those of age- and sex-matched nondemented controls [107,108]. Carboxyl-terminal-truncated fragments of apoE also appear to accumulate in NFTs in AD brains [107].
To determine whether expression of carboxyl-terminal-truncated apoE4 in transgenic mice induces AD-like neuropathological and behavioral changes, transgenic mouse lines expressing various levels of apoE4(Δ272–299) with its signal peptide in CNS neurons were established [108]. |
|
||
| Hippocampal or cortical neurons in these transgenic mice had numerous inclusion bodies containing phosphorylated tau that were positive for Gallyas silver staining, suggesting neurodegeneration. Staining with hematoxylin/eosin revealed degeneration of neurons expressing truncated apoE4. In the Morris water maze, the mice had learning and memory deficits. Thus, the carboxyl-terminal-truncated apoE4 is neurotoxic in vivo in transgenic mice and leads to AD-like neurodegeneration and behavioral deficit [108]. It is hypothesized that, in response to brain injury, gliosis, or Aβ neurotoxicity, neuronal apoE expression is induced or enhanced for purposes of repair or remodeling. However, these events trigger proteolytic processing of apoE4, resulting in fragments that are detrimental to repair and remodeling and lead to neurodegeneration (Figure 2) [47,48]. | |||
The amino-terminal 22-kDa thrombin-cleavage fragment (amino acids 1–191) of apoE4 seems to be more neurotoxic than the corresponding fragment of apoE3 [110,111]. This neurotoxicity may be mediated by lipoprotein receptors on the cell surface and by increased intracellular calcium levels [110,111]. However, apoE fragments generated in AD brains are different from the 22-kDa thrombin-cleavage fragment [107]. In fact, the lipid-binding domain (amino acids 241–272) is present in most of the fragments generated in AD brains [107], but not in the 22-kDa thrombin cleavage fragment, and is required for apoE fragments-related neurotoxicity in vitro and in vivo [107,108].
ApoE4 and its fragments stimulate tau phosphorylation and disrupts cytoskeletal structure and function
in vitro, apoE3 forms a sodium dodecyl sulfate–-stable complex with tau in a 1:1 ratio, whereas apoE4 does not interact significantly [112]. Phosphorylation of tau by a crude brain extract inhibits the interaction of apoE3 with tau [112], suggesting that apoE3 binds to nonphosphorylated tau. Furthermore, the amino-terminal domain of apoE3 is responsible for binding to tau [112]. In fact, apoE3 irreversibly binds to the microtubule-binding repeat regions of tau [113]. It has been shown that phosphorylation of tau increases in transgenic mice expressing human apoE4 in neurons but not in mice expressing apoE4 in astrocytes [109,114,115], indicating a neuron-specific effect of apoE4 on tau phosphorylation. The level of tau phosphorylation is also higher in apoE4 knock-in mice than apoE3 knock-in mice [116,117]. ApoE4-induced tau phosphorylation appears to be associated with Erk activation that can be modified by zinc concentration [118]. Furthermore, oxidative stress caused by folate and vitamin E deficiency exacerbates tau phosphorylation in apoE4, but not apoE3, transgenic mice, which can be prevented by S-adenosyl methionine supplementation [119].
Studies have suggested that carboxyl-terminal-truncated apoE4 stimulates tau phosphorylation and the formation of intracellular NFT-like inclusions in transgenic mice [108,109]. One target of the apoE4 fragments is the cytoskeletal components, such as tau and neurofilaments (Figure 2) [107].in vitro, the carboxyl-terminal-truncated fragments of apoE are toxic when expressed in neuronal cells, leading to the formation of cytoplasmic NFT-like inclusions in some cells [107]. Recently, the formation of NFT-like inclusions in neuronal cells expressing carboxyl-terminal-truncated apoE was confirmed, although the difference between truncated apoE3 and apoE4 was not quantified [120]. Thus, neurotoxicity induced by the apoE4 fragments might be related to cytoskeletal disruption.
ApoE4 and its fragments impair mitochondrial function
Mitochondrial dysfunction has been reported in AD, which is modulated by apoE genotype, with a greater effect in apoE4 than in apoE3 carriers [121-124,125]. In both AD patients and age-matched nondemented subjects, apoE4 is associated with decreased cerebral glucose metabolism [126-135], an effect that occurs decades before cognitive impairment become apparent [126,127,136] and, probably, also before significant Aβ deposition. Thus, apoE4 may cause mitochondrial dysfunction at very early stages in life. Mitochondrial dysfunction is clearly coupled with production of reactive oxygen species and increased oxidative damage, which in turn further impair mitochondrial activity. Temporary or sustained loss of mitochondrial function impairs cellular defenses and repair mechanisms, decreasing the ability of neurons to mount an appropriate stress response and causing cellular injury.
ApoE4 fragments target the mitochondria of neurons, leading to mitochondrial dysfunction and neurotoxicity (Figure 2) [138]. In cultured neuronal cells, the apoE4 fragment interacts with UQCRC2 and cytochrome C1, which are a component of respiratory complex III, and with COX IV 1, a member of complex IV [139]. Importantly, the receptor-binding region of apoE fragments is required for escape from the secretory pathway, and the lipid-binding region is required for mitochondrial interaction [138]. It appears that positively charged amino acids in the receptor-binding region, a feature shared among the protein translocation domains of many viral proteins [140,141], enable apoE4 fragments to translocate across membrane compartments of the secretory pathway and enter the cytosol, whereas the lipid-binding region interacts directly with the mitochondria [138]. Biophysical studies suggest that the lipid-binding domain within the C-terminal-truncated apoE4 has a less organized structure and greater exposure of the hydrophobic residues than full-length apoE4 [142,143], which might increase the interaction with mitochondrial membrane. Interestingly, almost 20 years ago, it was found that apoE avidly bound to mitochondrial ATPase [144]. In addition, small amounts of apoE have been identified in hepatocyte mitochondria [145]. Blocking the interaction of apoE4 fragments with mitochondria might prevent the detrimental effects on mitochondria function, and increasing the activity or number of mitochondria in neurons might also eliminate the detrimental effect of apoE4 fragments (Figure 2). Consistent with this possibility, treatment with rosiglitazone maleate, an insulin sensitizer and mitogenesis stimulator, appears to improve cognition in AD patients without apoE4 [137].
Time-lapse recordings of cultured neuronal cells demonstrate that apoE decreases mitochondrial mobility in an isoform-specific manner (apoE4 fragment > apoE4 > apoE3) (Jens Brodbeck and Yadong Huang, unpublished observations). Likewise, apoE4 impairs axonal transport of mitochondria in transgenic mice with neuron-specific expression of apoE4 [115]. Disruption of mitochondrial transport in neurons may contribute to AD pathology [146-149]. Moreover, regulation of mitochondrial distribution is essential for generating and maintaining distinct axonal and dendritic domains in response to local metabolic demand, morphological plasticity, and memory formation [150-154]. Failure to deliver mitochondria to appropriate sites results in energy starvation, local disruption of calcium homeostasis, and impairment of synapse formation [151,155,156].
Concluding remarks and potential therapeutic strategies
In addition to its well-documented Aβ-dependent roles [157], apoE4 has Aβ-independent roles in the pathogenesis of AD [43,47,48,158]. Independently of Aβ, apoE4 has detrimental effects on neuronal plasticity, undergoes aberrant proteolysis that generates neurotoxic fragments, stimulates tau phosphorylation and disrupts the cytoskeleton, and impairs mitochondrial function (Figure 3). In humans, the Aβ-dependent and Aβ-independent effects of apoE4 can act in concert to exacerbate the pathological and clinical phenotypes of AD.
Thus, in addition to developing anti-AD drugs that target Aβ-dependent roles of apoE4, drugs should also be designed to target Aβ-independent effects of apoE4. One potential strategy is to inhibit the apoE-cleaving enzyme that mediates apoE4 fragmentation. Another is to block the interaction of apoE4 fragments with cytoskeletal elements and mitochondria, thereby protecting against fragment-induced neurotoxicity. Another potential drug target is apoE4 domain interaction, which is responsible for apoE's susceptibility to proteolytic cleavage [43,158]. Small molecules could be designed to disrupt domain interaction by making apoE4 structurally and functionally more like apoE3 [42,43,158]. Clearly, new hope for effective therapeutics relies upon the ability of scientists to explore multiple lines of inquiry. It is certainly conceivable that there will be combination therapies, with drugs targeting Aβ itself and both Aβ-dependent and Aβ-independent effects of apoE4.